作者:师建军、孔磊、左小彪、刘登瑶、严蛟、冯志海
航天材料及工艺研究所机构先进功能复合材料技术国防科技重点实验室 北京 100076
通讯作者: 师建军, E-mail: shijj2016@yeah.net 冯志海, E-mail: fengzhh2006@sina.com
基金项目: 国家自然科学基金(基金号 51702076 基金 )和国家重点基础研究与发展计划项目(973计划,项目号 2015CB655200)资助
摘要: 通过Sol-Gel反应过程控制,调节酚醛树脂(PR)溶液和SiO2溶胶液的凝胶时间,共凝胶反应制备PR和SiO2凝胶网络互穿的PR/SiO2杂化气凝胶. 考察了杂化气凝胶表观密度、收缩率、孔结构、微观形貌、热稳定性和力学性能随SiO2气凝胶含量的基本变化规律. 研究结果表明:伴随SiO2气凝胶含量的增加,PR/SiO2杂化气凝胶表观密度正比增加;同纯PR有机气凝胶相比,杂化气凝胶孔结构得到明显改善,当[TEOS] = 1.50 mol/L时,平均孔径减小了一半,降至0.25 μm,比表面积由24.60 m2/g增至44 m2/g左右;微观形貌照片显示有机气凝胶网络骨架中的大孔逐渐被SiO2溶胶粒子填充,变成更细小疏松的孔结构形貌,孔径分布明显变宽,SiO2气凝胶在PR凝胶骨架中纳米尺度内均匀分散;无机SiO2气凝胶的引入能有效提高有机气凝胶骨架结构的强度和热稳定性,纳米分散SiO2溶胶粒子的热阻隔效应抑制了PR有机骨架的热分解速率,当[TEOS] = 1.00 mol/L时,杂化气凝胶的Tmax由539 °C提高到602 °C,热分解温度区间明显展宽.
气凝胶材料凭借自身低密度、低热导率、高比表面积、高孔隙率、纳米孔结构等诸多特性,在绝热、隔声、催化、电化学、吸附、分离等领域有着广泛的应用[1~3]. 无机气凝胶材料如SiO2、Al2O3气凝胶等具有极佳的高温隔热性能,在保温材料、高效催化剂载体、高温隔热、火箭低温燃料储箱、材料轻量化等领域有广泛的应用[4~6],但是无机气凝胶材料固有强度低、质脆的缺点成为航天材料应用过程中的瓶颈[7~9]. 有机气凝胶材料,如酚醛气凝胶、聚氨酯气凝胶、纤维素气凝胶等克服了无机气凝胶强度低、易碎裂的缺点,保留了低密度、高孔隙率、高比表面积、低热导的特性,聚合物凝胶网络骨架在提供强度的同时其分子链又可被功能化修饰和剪裁,赋予特定功能性. 然而,有机气凝胶耐温性有限,高温碳化后体积收缩严重,抗氧化性能不足,这些缺点大大限制了其在航天领域的应用范围[10~13].
随着气凝胶功能材料的不断开发以及应用范围的不断拓宽,单一有机或无机气凝胶材料已难以满足航天领域一些极端环境下的特殊功能需求,例如:在烧蚀防热复合材料中,单一聚合物气凝胶难以抵御高速燃气流的冲刷,形成的多孔碳化层极易被氧化. 因此,复合或杂化成为气凝胶材料应用中一个重要发展方向[14~16]. 此外,酚醛气凝胶作为碳气凝胶最重要的前驱体,其骨架结构强度、孔结构、抗氧化性等对碳气凝胶的性能有着重要影响. 在酚醛气凝胶骨架中引入无机抗氧化功能组元,能够改善气凝胶的结构强度,提高碳气凝胶的抗氧化性. 大量研究工作表明,通过溶胶-凝胶反应过程控制能够将SiO2、Al2O3、TiO2、CNT、石墨烯等无机组分成功引入到有机凝胶网络结构中,提高碳气凝胶的抗氧化性和耐高温性能[16~21]. 本研究基于普通酚醛树脂(PR)气凝胶报道了一种双体系凝胶网络结构杂化气凝胶,通过Sol-Gel反应过程控制和常压干燥的方法制备出PR/SiO2杂化气凝胶,以期改善酚醛气凝胶耐高温性、抗氧化性和力学强度.
1、实验部分
1.1、原材料
酚醛树脂(Phenolic Resin,PR),韩国科隆(KOLON)酚醛树脂HiRENOL®系列,牌号为KPH-F2003,Mw ≈ 2000;催化剂:六亚甲基四胺(HMTA),分析纯,国药集团化学试剂有限公司;正硅酸四乙酯,分析纯,国药集团化学试剂有限公司;无水乙醇,分析纯,西陇化工股份有限公司.
1.2、分析与测试
密度测试是将样品切割成规则的圆柱形状,用游标卡尺和电子分析天平分别准确测量计算其体积和质量,根据ρ = m/V计算表观密度.
孔结构通过美国康塔公司Poremaster GT60全自动孔隙度压汞分析仪测定,采用连续扫描模式,压力范围为0.007 ~ 206 MPa,室温测试.
微观形貌在英国CamScan公司Appolo300型场发射扫描电子显微镜(SEM)上进行表征,加速电压15 kV,观察前将样品进行喷金处理,元素分析通过SEM附带能谱仪(Oxford 7537型)进行表征.
SiO2的分散状态通过荷兰FEI公司G2F20S-TWIN高分辨透射电子显微镜进行分析,将气凝胶粉末分散在乙醇中,铜网捞起制样.
热重分析在美国Perkin-Elmer公司,Diamond TG/DTA热重-差热综合热分析仪上测定,氮气氛围,升温速率为20 K/min,温度扫描范围为室温至900 °C.
材料的压缩性能在美国美特斯万能试验机上进行,按照GB/T 1448-2005标准进行测试,压缩速率为5 mm/min,每组测试5个样品,取平均值.
1.3、双体系凝胶网络杂化气凝胶的制备
1.3.1、二氧化硅(SiO2)溶胶液的配制
称量一定量的正硅酸四乙酯(TEOS),加到无水乙醇溶剂中,配制成浓度为30 wt%的溶液,按照n(TEOS):n(H2O) = 1:6,c(HNO3) = 1.0 × 10−3 mol/L的计量比,分别滴加去离子水和0.1 mol/L的稀硝酸溶液,室温下搅拌反应30 min.
1.3.2、双体系凝胶网络结构杂化气凝胶的制备
称量已配制的30 wt%二氧化硅溶胶液放入烧杯中,按照计量比加入酚醛树脂(PR)和催化剂(HMTA),在保持PR浓度为20 wt%的条件下补加一定量的无水乙醇溶剂,搅拌均匀,直至酚醛树脂完全溶解,得到杂化溶胶液体系. 其中催化剂的浓度为0.25 mol/L,杂化溶胶液体系中二氧化硅溶胶液加入量以单体TEOS的摩尔浓度计量,分别为[TEOS] = 0.25、0.5、1和1.5 mol/L. 将上述杂化溶胶液转移至模具中,密封,在140 °C下Sol-Gel反应6 h,形成稳定的双体系凝胶网络结构,接着在80 °C下继续陈化80 h. 反应完毕后,从模具中取出杂化湿凝胶,自然晾置24 h,然后在45 °C烘箱中常压干燥24 h,最终得到PR/SiO2双体系凝胶网络结构杂化气凝胶(HPA).
2、结果与讨论
2.1、双体系凝胶网络结构杂化气凝胶的制备
Sol-Gel技术是制备气凝胶材料最常用的手段,PR/SiO2杂化气凝胶(HPA)制备过程的关键是如何控制各组分间的化学反应过程和调节凝胶时间,使得2种体系具有相近的凝胶时间,保证在后续进一步凝胶化的过程中不发生严重相分离,共凝胶反应形成双体系凝胶网络杂化气凝胶. SiO2气凝胶是在强酸催化作用下通过TEOS的水解缩合形成Si―O―Si的交联网络结构,室温条件下反应速率相对较大,凝胶时间短;酚醛树脂溶液体系中,加入弱碱性催化剂后,聚合物溶液体系呈弱碱性,室温下反应速率相对低,凝胶时间长. 为了制备具有均相结构的PR/SiO2杂化气凝胶,减小混合溶液体系中弱碱性催化剂对SiO2溶胶体系TEOS单体水解、缩合反应的影响,首先将TEOS单体在强酸催化条件下进行凝胶化反应,形成含有低交联度Si―O―Si网络结构的前驱体溶胶. 在混合溶胶液体系中,为增加PR溶液的Sol-Gel反应速率,缩短凝胶时间以匹配SiO2溶胶体系,利用TEOS水解缩合和酚醛树脂交联反应对温度敏感性的差异,采用提高初始反应温度(140 °C)的方法. 待PR溶胶粒子和SiO2溶胶粒子形成稳定的互穿凝胶网络结构后,在80 °C下进一步陈化,形成以有机PR凝胶网络为骨架无机SiO2溶胶粒子穿插的杂化湿凝胶. 通过控制Sol-Gel反应过程,调节杂化气凝胶的孔结构,减小干燥过程中毛细收缩力对气凝胶骨架结构的影响,实现常压干燥[22],得到块状双体系凝胶网络结构PR/SiO2杂化气凝胶,制备过程如图1所示.
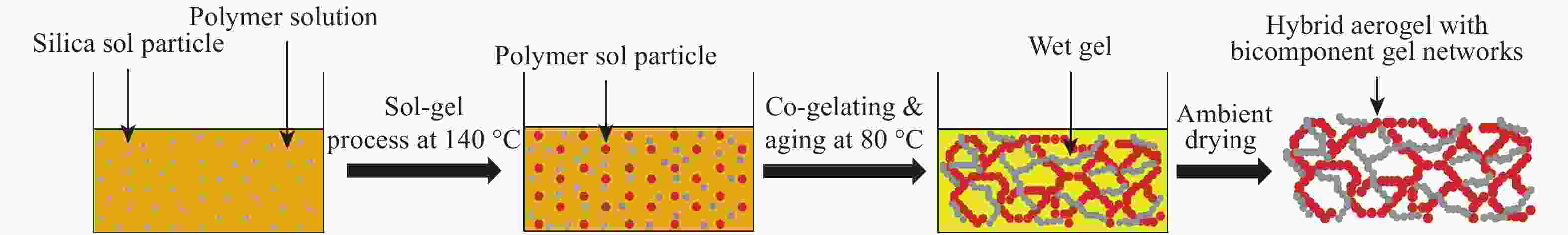
Figure 1. Schematic preparation of PR/SiO2 hybrid phenolic aerogels with bi-component gel networks
图2为PR/SiO2杂化气凝胶的傅里叶红外光谱(FTIR). FTIR显示通过共凝胶反应引入SiO2气凝胶后,杂化气凝胶在3300 ~ 3400 cm−1之间出现馒头形鼓包,对应于Si―OH伸缩振动特征峰,962 cm−1处出现Si―OH的面外摇摆振动特征峰. 在1052 和793 cm−1处出现Si―O―Si骨架的不对称和对称收缩特征峰. 随着TEOS浓度的增加,SiO2气凝胶的含量相应增加,对应官能团红外特征峰的强度也渐渐变强,而PR有机骨架在1300 ~ 1700 cm−1之间对应的苯环特征峰几乎保持不变. FTIR谱图分析结果表明:SiO2凝胶网络结构成功地穿插入PR有机气凝胶骨架中,随着TEOS浓度的增加,SiO2气凝胶含量成正比增加,并且PR有机气凝胶的骨架结构未被破坏.
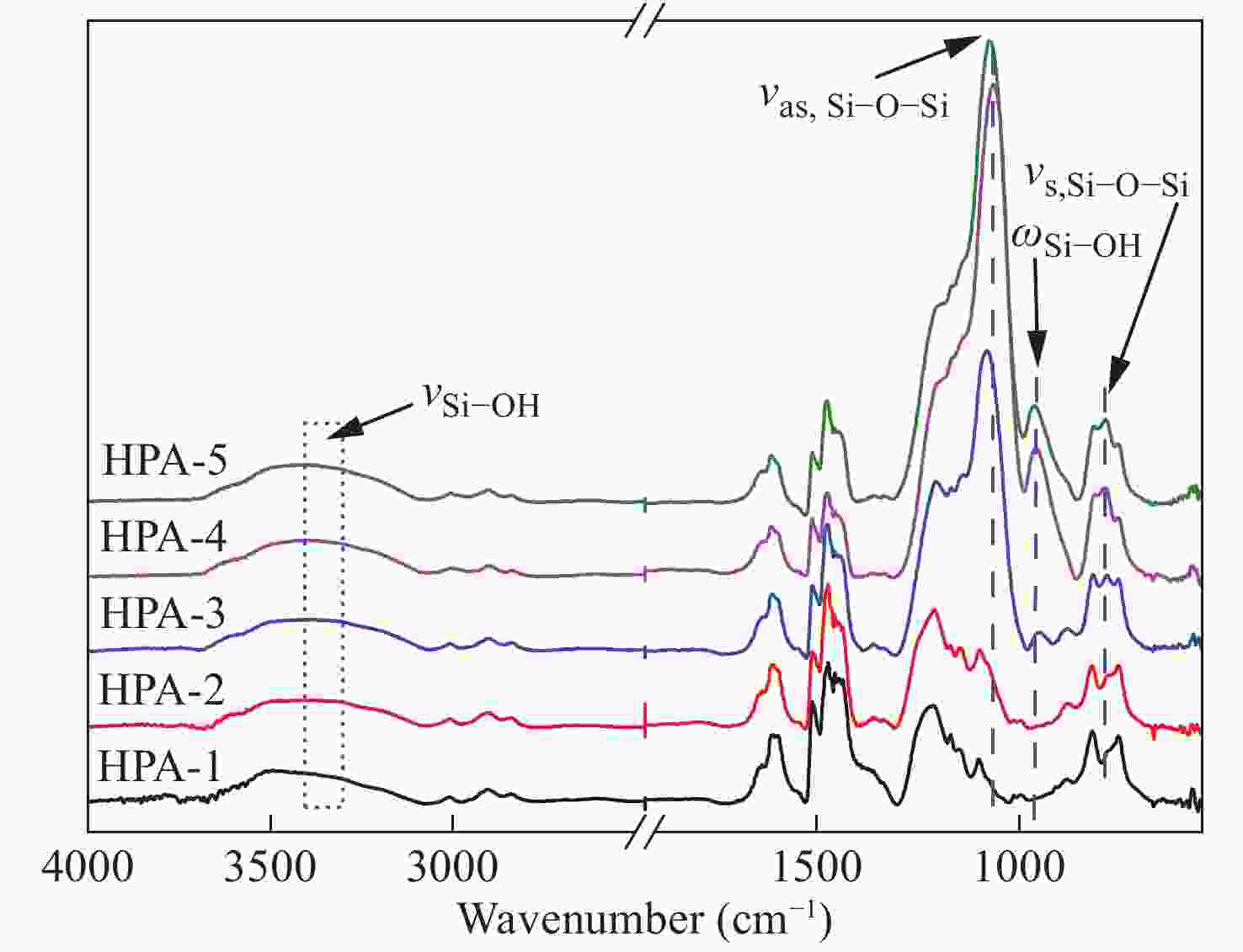
Figure 2. FTIR spectra of PR/SiO2 hybrid phenolic aerogels (HPA) with different SiO2 aerogel contents
表1为共凝胶反应制备得到的PR/SiO2杂化气凝胶的基本性能数据. 可以看出,随着TEOS单体浓度的升高,杂化气凝胶的表观密度增加,线收缩率、平均孔径和比孔容减小. 在溶胶-凝胶反应过程中,随着TEOS浓度的提高,穿插在有机气凝胶骨架中的SiO2溶胶粒子增加,提高了有机气凝胶的骨架强度,能够抵御干燥过程中由毛细作用而产生的收缩应力,使得收缩率降低. SiO2溶胶粒子通过Sol-Gel化学反应同PR有机溶胶粒子共凝胶作用穿插交织,形成杂化凝胶网络,无机和有机溶胶粒子在物理和化学作用力下紧密吸附、溶合,纯PR有机气凝胶中原有的较大孔隙被SiO2溶胶粒子填充,形成更细小的孔隙结构(图3),从而造成PR/SiO2气凝胶平均孔径和比孔容减小,比表面积显著增加,孔隙率基本不变. 表1中孔结构分析数据显示,PR有机气凝胶的平均孔径、比孔容和比表面积分别为0.55 μm、3.39 cm3/g和24.6 m2/g,当引入少量SiO2气凝胶时,杂化气凝胶的平均孔径和比孔容分别降至0.36 μm和3.24 cm3/g,比表面积增大至35.74 m2/g;当[TEOS] = 1.50 mol/L时,平均孔径降低至0.25 μm,比孔容降至PR有机气凝胶的45%左右,比表面积增加为PR气凝胶的1.8倍.

Figure 3. Micromorphological images of (a) PR aerogel and (b) PR/SiO2 hybrid aerogel with TEOS monomer concentration of 1.00 mol/L
| Sample | [TEOS] (mol/L) |
ρ a (g/cm3) |
LS b (%) |
D c (μm) |
V sc (cm3/g) |
S c (m2/g) |
Pc <italic /> (%) |
Td5d (°C) |
Tmaxe (°C) |
Residue (900 °C, %) |
| HPA-1 | 0.00 | 0.241 | 17.67 | 0.55 | 3.39 | 24.60 | 96.85 | 206.5 | 538.8 | 48.6 |
| HPA-2 | 0.25 | 0.257 | 16.96 | 0.36 | 3.24 | 35.74 | 96.64 | 196.3 | 566.0 | 51.4 |
| HPA-3 | 0.50 | 0.262 | 16.70 | 0.31 | 3.09 | 43.48 | 96.48 | 141.0 | 563.5 | 58.5 |
| HPA-4 | 1.00 | 0.281 | 16.02 | 0.28 | 2.74 | 38.51 | 95.99 | 105.0 | 602.1 | 61.1 |
| HPA-5 | 1.50 | 0.310 | 14.54 | 0.25 | 2.72 | 43.94 | 96.43 | 78.0 | 566.6 | 62.3 |
| a Apparent density of aerogel calculated by ρ = m/V; b LS refers to linear shrinkage of aerogel calculated by (D1 − D0) × 100/D0. D1, D0 are the diameters of wet and dry aerogels respectively; c From the measurement of mercury porosimerty according to GB/T 21650.1-2008; d The temperature corresponds to 5% weight loss of the materials; e The temperature corresponds to the maximum pyrolysis rate of the materials | ||||||||||
Table 1. Results and basic properties of PR/SiO2 hybrid phenolic aerogels with bi-component gel networks
2.2、SiO2气凝胶对杂化气凝胶孔结构的影响
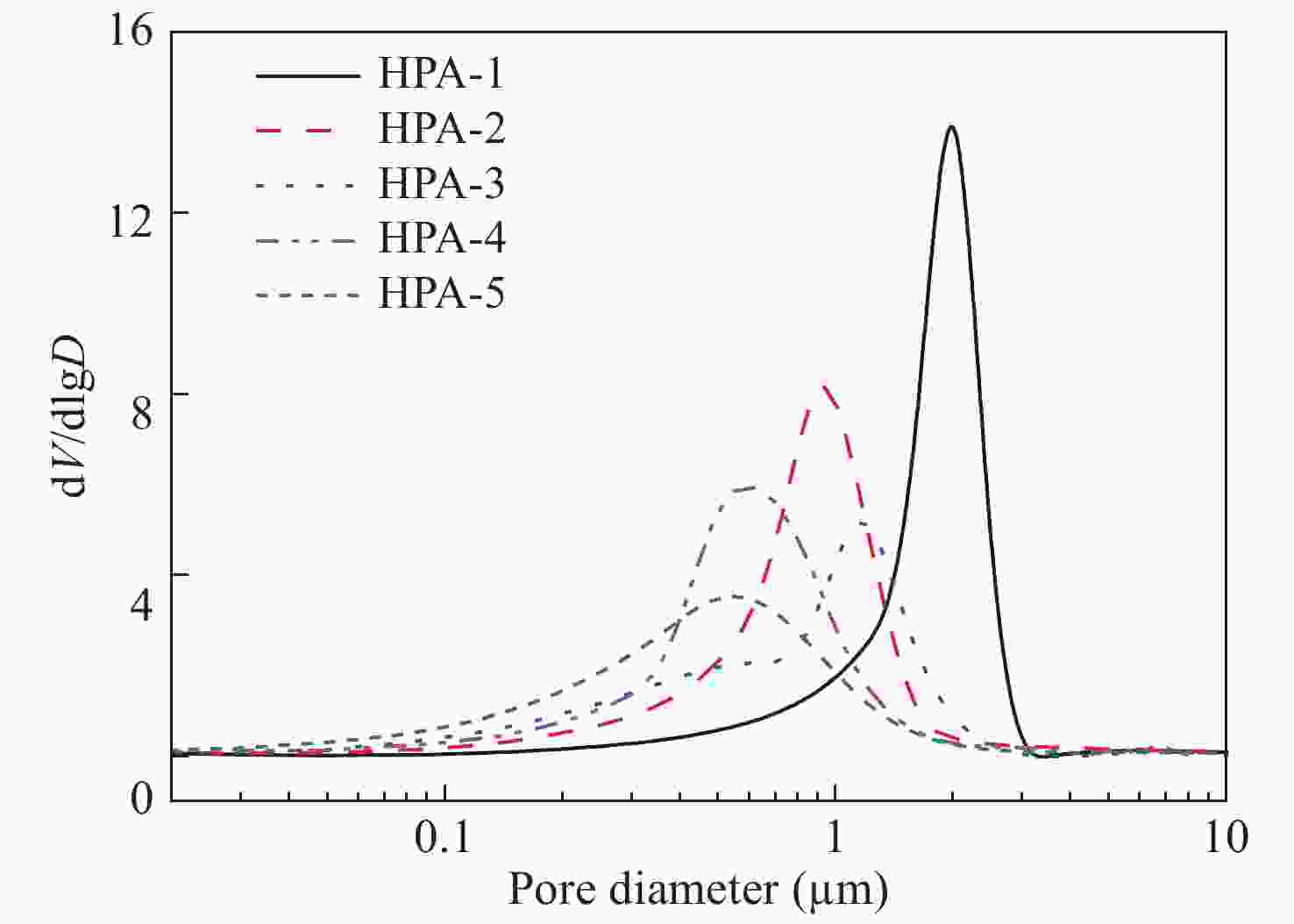
Figure 4. Pore diameter and its distribution curves of PR/SiO2 hybrid phenolic aerogels with different SiO2 aerogel content
从表1 PR/SiO2气凝胶孔结构数据和图4孔径分布曲线可以看出:酚醛有机骨架中无机SiO2气凝胶的引入对PR/SiO2杂化气凝胶的孔结构有显著影响. 在共凝胶反应形成双体系凝胶网络结构过程中,SiO2溶胶粒子沿着PR有机气凝胶骨架通过物理吸附或化学交联形成互穿的杂化凝胶网络,原PR有机气凝胶的大孔逐渐被SiO2溶胶粒子填充(图3所示). 当加入TEOS单体浓度[TEOS] = 0.25 mol/L时,HPA-2的孔径分布曲线的最可几孔径(Dp)向小孔径方向移动,孔径分布变宽;当[TEOS] = 0.50 mol/L时,孔径分布曲线出现双峰,Dp ≈ 0.5 μm处出现的宽泛峰主要由SiO2次级溶胶粒子的堆砌引起,Dp ≈ 1 μm处的峰反映出酚醛有机骨架结构自身的孔结构. 随着TEOS浓度的增加([TEOS] = 1.00和1.50 mol/L),SiO2溶胶粒子对PR气凝胶骨架孔径分布的影响占主导,PR/SiO2杂化气凝胶的孔径急剧降低,分布变宽. 表1数据结果显示当[TEOS] = 1.50 mol/L时,气凝胶的平均孔径降低至250 nm左右,孔径分布在40 nm ~ 2.5 μm之间. 图3直观地显示出PR有机气凝胶骨架中引入SiO2气凝胶后的微观孔结构形貌,可以看出PR气凝胶次级溶胶粒子直径在100 ~ 200 nm之间,相互堆砌聚集构成分型孔结构. 在PR有机骨架结构中引入SiO2气凝胶后,溶胶粒子变得细小,直径减小至50 nm左右,原有骨架结构中的大孔结构被填充,形成更细小疏松的孔结构形貌. 孔结构和微观形貌结果表明:通过调节TEOS单体浓度来控制引入SiO2气凝胶的含量,能够调控杂化气凝胶的微观孔结构形貌,进而调节PR/SiO2气凝胶的宏观性能.
2.3、双体系凝胶网络结构杂化气凝胶的微观形貌结构
图5为不同TEOS单体浓度下的PR/SiO2杂化气凝胶微观形貌的SEM照片. 可以看到随着TEOS单体浓度的增加,PR有机气凝胶骨架的大孔逐渐被SiO2溶胶粒子填充,形成互穿结构的杂化凝胶网络,表面形貌由疏松的大孔结构逐步向细碎、疏松的小孔结构转变. 图5(a)为纯PR有机气凝胶的SEM照片,次级溶胶粒子为规则的球形,直径在200 nm左右. 随着SiO2溶胶粒子含量的增加(图5(c) ~ 5(e)),杂化溶胶粒子逐步变得不规则,粒子之间结合更为紧密,互相“溶合”在一起. 共凝胶反应制备PR/SiO2杂化气凝胶时,高温条件下酚醛聚合物分子链交联反应速率大[23~25],而对TEOS水解缩合反应速率影响不大[26~28],此时PR有机溶胶粒子快速形成、聚集、长大,形成有机凝胶网络结构[29]. TEOS单体不断进行水解,形成的SiO2溶胶粒子在纳米粒子集聚效应下,倾向于吸附有机凝胶网络生长、聚集和溶合,形成SiO2凝胶网络,整个过程如图1所示意. 2种凝胶网络相互穿插、堆积,形成双体系凝胶网络结构,PR有机骨架结构中大孔被SiO2溶胶粒子填充,形成更细小的孔结构,比表面积明显增加,而孔隙率几乎保持不变.


2.4、SiO2气凝胶对杂化气凝胶热稳定性的影响
图7为PR/SiO2杂化气凝胶在氮气氛围下的热失重分析曲线(TGA)和微分曲线(DTG),PR有机气凝胶引入SiO2气凝胶后在100 °C附近出现一个明显的失重过程,并且随着TEOS单体浓度的增加失重越发明显. 从表1 TGA数据读出,随着TEOS单体浓度的增加,杂化气凝胶失重5%所对应的温度Td5逐渐降低,当[TEOS] = 1.50 mol/L时降到78 °C. 这一失重主要来源于气凝胶中吸附的小分子物质(CO2、H2O、空气等)的物理挥发和SiO2凝胶网络结构中未完全缩合Si―OH之间的脱水缩合反应[30, 31]. 随着TEOS加入量的增加,未完全缩合Si―OH含量增加,进一步造成失重. 杂化气凝胶在100 ~ 300 °C之间还有一个轻微失重过程,源于酚羟基分子间的脱水和交联网络中三苄胺的分解[22, 32, 33].
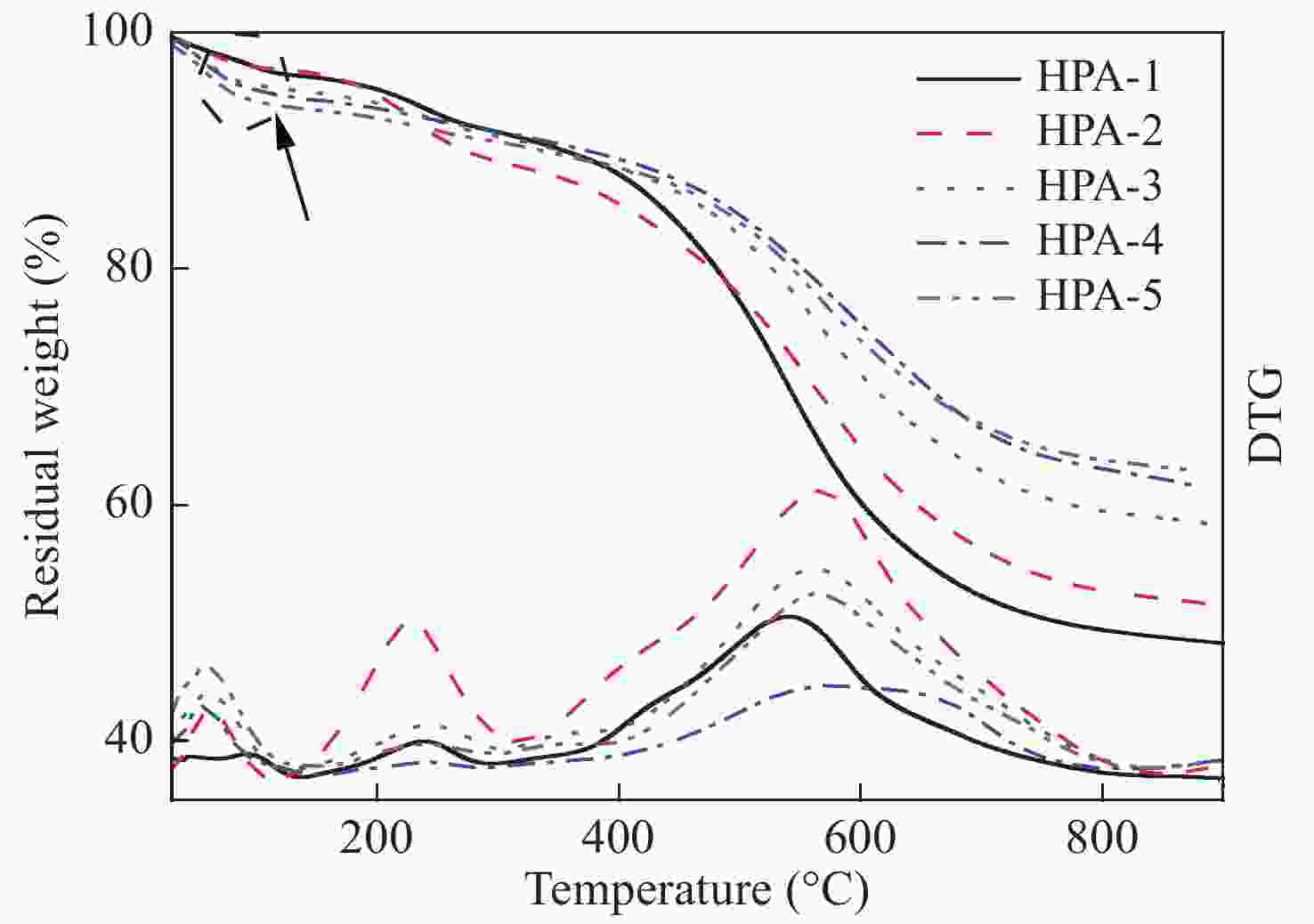
从表1和图7也可以看出,PR有机气凝胶骨架引入SiO2气凝胶后,最大热分解速率所对应的温度Tmax (DTG曲线峰值温度)和残留率均有大幅提高. 图7显示纯PR气凝胶的Tmax为539 °C左右,残碳率约为48.6%,当[TEOS] = 0.25 mol/L时,PR/SiO2杂化气凝胶的Tmax增加到566 °C左右,残留率增长至51.4%. 当[TEOS] > 0.25 mol/L时, Tmax均保持在560 °C以上,杂化气凝胶的残留率成正比增加,当[TEOS] = 1.50 mol/L时,900 °C残重达到62.3%. 此外,有机气凝胶骨架引入SiO2气凝胶后,最大热分解速率减小,热分解产物释放速率受到抑制,TGA和DTG曲线中表现为热分解温度区间变宽. 从图7中看出PR有机气凝胶的热分解温度区间在380 ~ 690 °C之间,而引入SiO2气凝胶后热分解温度区间在320 ~ 800 °C之间,尤其在[TEOS] = 1.00 mol/L时,DTG曲线峰由尖锐变得宽泛化. 这主要是因为高比表面积的酚醛气凝胶网络结构被无机SiO2溶胶粒子吸附、包裹和溶合,纳米分散的SiO2溶胶粒子起到热阻隔的作用,有效抑制酚醛分子链的降解,延缓有机气凝胶骨架的热分解过程;同时,纳米分散的SiO2溶胶粒子在高温碳化后形成的无机SiO2颗粒均匀分散于热解碳骨架中,提高杂化气凝胶的残留率. 据此可以推断:通过调控有机气凝胶网络骨架中SiO2气凝胶的含量和相分散状态,能够提高酚醛有机气凝胶的热稳定性,增加PR/SiO2杂化气凝胶碳化后的骨架强度.
2.5、双体系凝胶网络结构杂化气凝胶的压缩性能
表2为PR/SiO2杂化气凝胶的压缩性能数据,PR有机气凝胶骨架引入SiO2气凝胶后压缩强度和压缩模量均显著增加,随着TEOS浓度的增大,压缩强度成正比增加. 当TEOS浓度为1.50 mol/L时,杂化气凝胶的压缩强度达到2.5 MPa左右,与纯PR气凝胶样品(HPA-1)相比增加了1倍左右,压缩模量达到44.65 MPa. 图8中压缩应力-应变曲线显示,当TEOS单体浓度大于0.50 mol/L时,杂化气凝胶的韧性增大,压缩应变由原来的10%左右增加到20%以上. 杂化气凝胶压缩性能的提高主要得益于SiO2纳米溶胶粒子的引入,SiO2气凝胶网络与PR有机气凝胶网络相互穿插、吸附和溶合,SiO2和PR溶胶粒子紧密结合,刚性的无机溶胶粒子既能够增加有机气凝胶的骨架结构强度,在压缩破坏过程中又能起到润滑作用,增加材料的韧性.
| Sample | [TEOS] (mol/L) | Compressive strength (MPa) | Compressive modulus (MPa) |
| HPA-1 | 0.00 | 1.16 ± 0.06 | 33.75 ± 2.70 |
| HPA-2 | 0.25 | 1.32 ± 0.02 | 38.30 ± 4.10 |
| HPA-3 | 0.50 | 1.42 ± 0.17 | 39.30 ± 6.20 |
| HPA-4 | 1.00 | 2.30 ± 0.05 | 37.30 ± 3.00 |
| HPA-5 | 1.50 | 2.50 ± 0.27 | 44.65 ± 4.41 |
Table 2. Compressive properties of PR/SiO2 hybrid phenolic aerogels with different SiO2 aerogel content
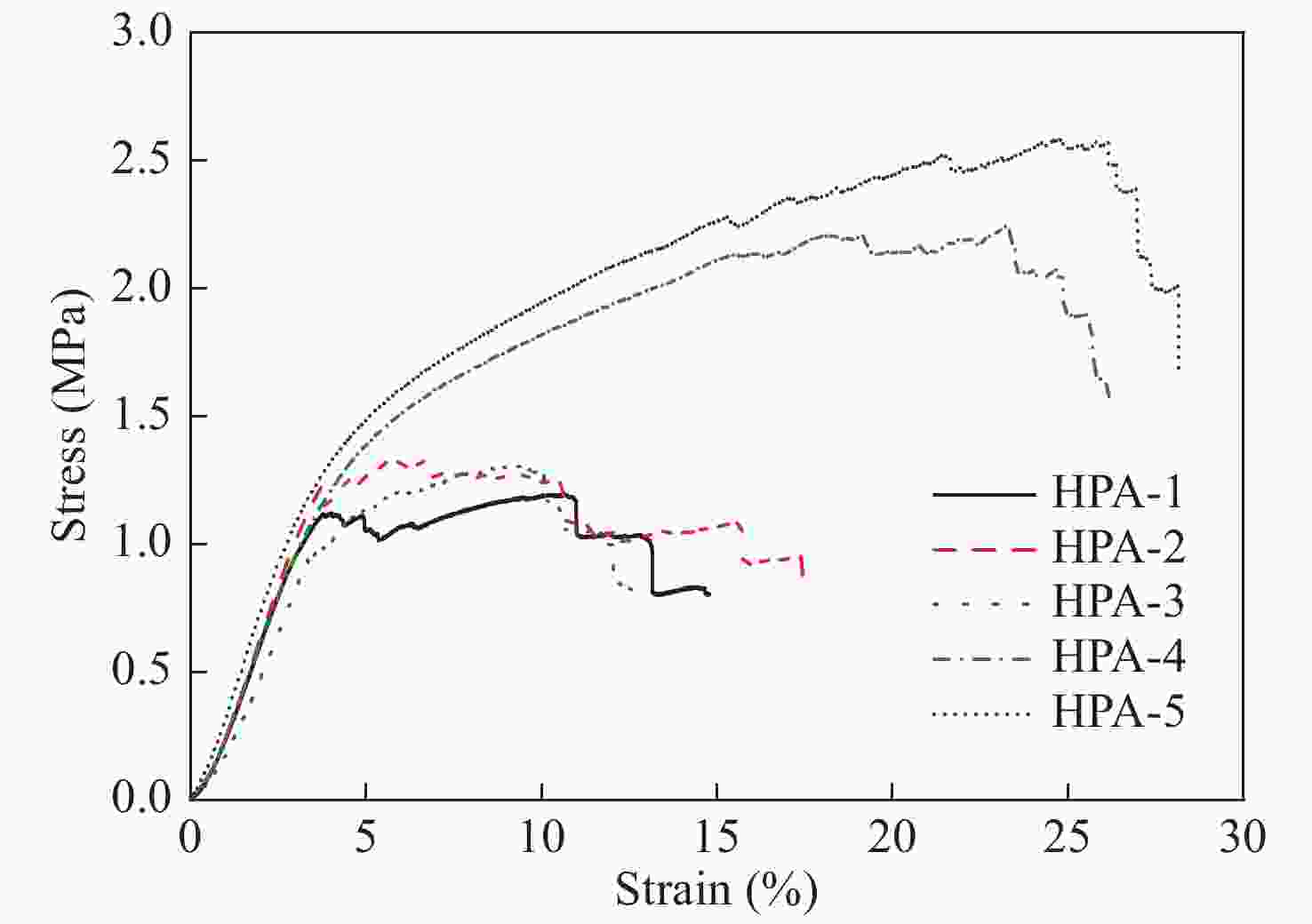
Figure 8. Stress versus strain plots of PR/SiO2 hybrid phenolic aerogels
3、结论
通过对二氧化硅溶胶和酚醛树脂溶液凝胶时间的调控,共凝胶反应制备出PR/SiO2杂化气凝胶. FTIR表征、SEM元素面扫描和TEM结果显示SiO2被成功引入到PR有机气凝胶网络结构,并且呈纳米级分散. 随着SiO2气凝胶引入量的增加,杂化气凝胶的表观密度成正比增加,线收缩率减小,杂化气凝胶比表面积增加,平均孔径减小,同时保持着高孔隙率(96%左右). 当TEOS单体浓度达到1.50 mol/L时,平均孔径减小到纯PR气凝胶的45%,达到250 nm,比表面积增加了80%左右. 无机SiO2气凝胶纳米溶胶粒子的引入,使酚醛气凝胶基体中的大孔被填充,聚合物凝胶网络被纳米SiO2溶胶粒子包覆,形成更细小的孔隙结构. SiO2气凝胶对孔结构和微观形貌结构的改善,有效地提高了PR气凝胶网络结构的强度和热稳定性,纳米分散SiO2溶胶粒子的热阻隔效应有效抑制有机聚合物骨架的热分解,延缓酚醛分子链的热分解速率,拓宽了热分解温度区间.
参考文献
- [1]Fricke J, Emmerling A. J Sol-Gel Sci Technol, 1998, 13(1-3): 299 − 303
- [2]Aegerter M A, Nicholas L, Matthias M M. Aerogels Handbook. New York: Springer-Verlag New York, 2011. 932
- [3]Fathy N A, Attia A A, Hegazi B. Desalin Water Treat, 2016, 57(21): 9957 − 9970
- [4]Niimi R, Kadono T, Tsuchiyama A, Okudaira K, Hasegawa S, Tabata M, Watanabe T, Yagishita M, Machii N, Nakamura A M, Uesugi K, Takeuchi A, Nakano T. Astrophys J, 2012, 744(1): 18 − 22
- [5]Baetens R, Jelle B P, Gustavsen A. Energ Buildings, 2011, 43(4): 761 − 769
- [6]Ren H, Wu D, Li J, Wu W. Mater Des, 2018, 140: 376 − 386
- [7]Wang J, Kuhn J, Lu X. J Non-Cryst Solids, 1995, 186: 296 − 300
- [8]Shimizu T, Kanamori K, Nakanishi K. Chem Eur J, 2017, 23(22): 1 − 13
- [9]Boday D J, Loy D A. J Non-Cryst Solids, 2015, 427: 114 − 119
- [10]Ghafoorian N S, Bahramian A R, Seraji M M. Mater Design, 2015, 86: 279 − 288
- [11]Qian H, Kucernak A R, Greenhalgh E S, Bismarck A, Shaffer M S P. ACS Appl Mater Interfaces, 2013, 5(13): 6113 − 6122
- [12]Bessire B K, Lahankar S A, Minton T K. ACS Appl Mater Interfaces, 2015, 7(3): 1383 − 1395
- [13]Bahramian A. Iran Polym J, 2013, 22(8): 579 − 589
- [14]He S, Bi Y, Zhang Y, Cao H, Shi X, Luo X, Zhang L. J Sol-Gel Sci Technol, 2015, 74(1): 175 − 180
- [15]Mazraeh-Shahi Z T, Shoushtari A M, Bahramian A R. Procedia Mater Sci, 2015, 11: 571 − 575
- [16]Kong Y, Zhong Y, Shen X, Cui S, Yang M, Teng K, Zhang J. J Non-Cryst Solids, 2012, 358(23): 3150 − 3155
- [17]Seraji M M, Ghafoorian N S, Bahramian A R, Alahbakhsh A. J Non-Cryst Solids, 2015, 425: 146 − 152
- [18]Yin R, Cheng H, Hong C, Zhang X. Compos Part A: Appl S, 2017, 101: 500 − 510
- [19]Kong Y, Shen X, Cui S, Fan M. Ceram Int, 2014, 40(6): 8265 − 8271
- [20]Wu X, Li W, Shao G, Shen X, Cui S, Zhou J, Wei Y, Chen X. Ceram Int, 2017, 43(5): 4188 − 4196
- [21]Gao X, Lv H, Li Z, Xu Q, Liu H, Wang Y, Xia Y. RSC Adv, 2016, 6(109): 107278 − 107285
- [22]师建军), Yan Jiao(严蛟), Kong Lei(孔磊), Yang Yunhua(杨云华), Zuo Xiaobiao(左小彪), Feng Zhihai(冯志海), Yu Ruilian(余瑞莲). Acta Polymerica Sinica(高分子学报), 2016, (2): 179 − 186
Shi Jianjun - [23]葛铁军), Yang Xue(杨雪), Li Yingming(李英明). J Shenyang Univ Chem Technol(沈阳化工大学学报), 2013, 27(4): 327 − 332
Ge Tiejun - [24]Carotenuto G, Nicolais L. J Appl Polym Sci, 1999, 74(11): 2703 − 2715
- [25]He G, Riedl B, Aït-Kadi A. J Appl Polym Sci, 2003, 87(3): 433 − 440
- [26]Kamiya K, Yoko T, Suzuki H. J Non-Cryst Solids, 1987, 93(2): 407 − 414
- [27]Colby M W, Osaka A, Mackenzie J D. J Non-Cryst Solids, 1988, 99(1): 129 − 139
- [28]Colby M W, Osaka A, Mackenzie J D. J Non-Cryst Solids, 1986, 82(1): 37 − 41
- [29]Hench L L, West J K. Chem Rev, 1990, 90(1): 33 − 72
- [30]Rao A V, Kulkarni M M, Amalnerkar D P, Seth T. Appl Surf Sci, 2003, 206(1): 262 − 270
- [31]Rao A P, Rao A V, Pajonk G M. Appl Surf Sci, 2007, 253(14): 6032 − 6040
- [32]Jiang H, Wang J, Wu S, Yuan Z, Hu Z, Wu R, Liu Q. Polym Degrad Stab, 2012, 97(8): 1527 − 1533
- [33]Chen Y, Chen Z, Xiao S, Liu H. Thermochim Acta, 2008, 476(1-2): 39 − 43
相关新闻
-

铜亮相柔性导体领域
由铜制成的传感器具有价格低、重量轻、易弯曲和强导电性的特点。弯曲、拉伸、扭曲、折叠——现代材料的轻便、柔韧和高导电率等特点使其具有非凡潜力,例如制造人造皮肤和电子纸。想让这些概念技术变成现实仍需时日,不过,一种基于纳米铜丝及PVA(聚乙烯醇)纳米胶水的新方法可能成为救星。“气凝胶巨石”(aerogel monoliths)作为超轻领域的一个成功发明,在很大程度上依赖于珍贵的纳米金丝和银丝。取而代之,莫纳什大学及墨尔本纳米制造中心的研究人员利用廉价的铜降低了柔性导体的成本,使其更具商业价值。“气凝胶巨石就像厨房里的海绵,只是我们的导体是由超精细的纳米铜丝构成,并且利用了一个叫做冷冻干燥的制造工序
-
气凝胶材料在LNG液化天然气方面的应用
在LNG工程及其他低温项目建设中,低温、超低温设备涉及到的温度大都在-40至-170摄氏度。最常用的深冷保冷材料主要有PUR/PIR,发泡玻璃,橡塑,改性酚醛泡沫等,这些材料较之于先前使用的珍珠岩材料,无论从性能还是施工方面看,都有了很大的改善。保冷效果的好坏不仅关系到整个设备的输送效率,而且对装置的安全生产也有至关重要的影响。合适的保冷材料不仅能够降低能耗、减少冷量损失,而且为符合环保要求、为企业安全生产和创造更好的效益提供了保障。气凝胶型保温材料的出现,似乎正是为了这种深冷型保温而量身定做,…
-
气凝胶采光玻璃特性介绍
保温性能——气凝胶采光玻璃热工性能 气凝胶采光玻璃的热工性能稳定,不受安装角度的影响 能耗模拟——不同类型玻璃窗整窗U值 Optics,Window7.6,RESFEN6由美国劳伦斯伯克利国家实验开发的专门计算门窗及幕墙的热工性能的软件。模拟计算采用美国波斯顿非典型年气候文件,建筑类型为普通2层楼住宅,其中玻璃窗总面积占建筑面积42%, 模拟结果  …
-
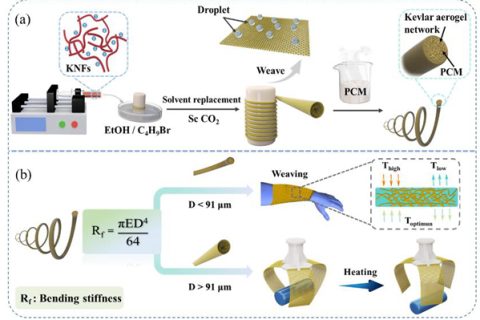
弯曲刚度导向策略制备Kevlar气凝胶限域的有机相变纤维研究获进展
智能或功能纤维在可穿戴及其他高科技领域已显示出较大潜力,但设计和制备结构可控的智能或功能纤维仍然面临挑战。最近,中国科学院苏州纳米技术与纳米仿生研究所气凝胶团队通过一种弯曲刚度导向策略,制造出具有不同功能的有机相变纤维,并探索了其在不同领域的应用前景(图1)。首先,研究利用正溴丁烷/乙醇混合溶剂作为湿法纺丝的凝固浴,对Kevlar纳米纤维(KNF)质子化同时进行疏水功能化,再通过超临界干燥,制备出疏水的凯夫拉气凝胶纤维(H-KAFs);接着,以H-KAFs为载体,使有机相变材料石蜡(PW)限域于…
-
纳米纤维素制备柔性体相空气电极材料研究取得新进展
纤维素是自然界中广泛分布、含量丰富的天然高分子材料,在制浆造纸产业中作为重要的材料,其应用领域不仅仅用于生产纸张、功能纸, 也可以开发纳米纤维素及其深度应用。近年来,来源于纤维素的纳米纤维素作为一种可再生及环境友好的纳米材料受到了众多科研人员的关注,不仅拥有纤维素的基本特征,还具备了纳米材料的典型的特征,如质轻、较高的表面活性、比表面积大、杨氏模量高、吸附能力强和反应活性高等,赋予了纳米纤维素独特的光学性能、流变性能和机械性能,在制浆造纸中具有广阔的应用前景,如纸浆的增强、细小纤维和填料的助留等…
-
气凝胶在LNG储运应用中的优点浅析
LNG是即液化天然气(liquefied natural gas)的缩写(具体可以百科查询)。在LNG储藏和运输中涉及到低温项目建设和深冷保温运输设备,一般要求温度大约在-40℃至-170℃。目前常用的深冷保冷材料主要有PUR/PIR、发泡玻璃、橡塑、酚醛泡沫等,这些保温材料相比以前使用的珍珠岩保温材料,从性能及施工方面,都有了很大的改善。但上述保温材料在使用过程中,还出现了如下缺陷: 保温性能衰减很快,维护成本高。 保冷效果差,冷损耗大,容易给天然气或其他压缩气体的储藏运输带来危险。 包裹厚度…