

在合成有机分子和天然产物中,结构的确定是一项非常具有挑战性的工作。结构上接近的异构体和非对映异构体在1D NMR光谱中的差异非常细微,要想区分它们得耗费大量的时间和精力。
利用计算机进行核磁谱图识别给研究者提供了大量帮助,其原理是基于密度泛函理论(DFT)计算所有不确定结构非对映异构体的核磁位移,并使用相关系数、平均绝对误差(MAE)和校正平均绝对误差(CMAE)等参数将这些预测结果与已公布的光谱数据进行比较。其中,DP4分析是一种特别强大的工具,它不仅可以预测分子的立构化学特性,还可以给出每个可能结构是否正确的概率,在天然产物及药物合成中已有成功应用。
自发布以来,DP4的计算过程已经进行了极大的简化,用户输入越来越少。但是,最耗费用户精力的仍然是NMR谱图的归属问题,这不仅非常耗时费力,而且容易出错。
少数商用软件,如Mestrelab Mnova,虽然为1H NMR谱图提供了归属算法,但无法对原始NMR数据进行自动处理和归属。
成果介绍
基于以上分析,剑桥大学Jonathan M. Goodman教授课题组针对1H和13C NMR原始数据,提出了一种谱图自动处理和归属方法DP4-AI,它可以自动进行有机分子立构化学特性和结构歧义的预测。研究发现NMR-AI可以在1分钟左右的时间处理完NNR原始数据,而此前同样的任务大约需要8个小时,速率提升了480倍,每天可以处理的分子数量增加了60倍,这使得高通量NMR谱图分析成为可能,为通过机器学习发现新的分子结构铺平了道路。

DP4-AI的结构和计算流程
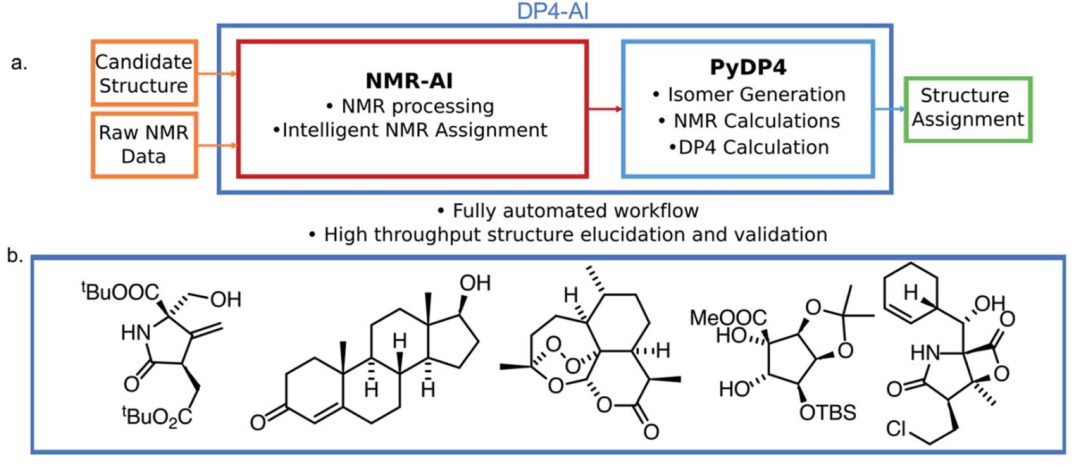
DP4-AI包含了NMR-AI和PyDP4两部分,其中NMR-AI负责处理用户输入的NMR原始数据,并进行化学位移的归属,PyDP4则对这一归属正确与否的概率进行计算,从而自动阐述分子的立体化学结构。
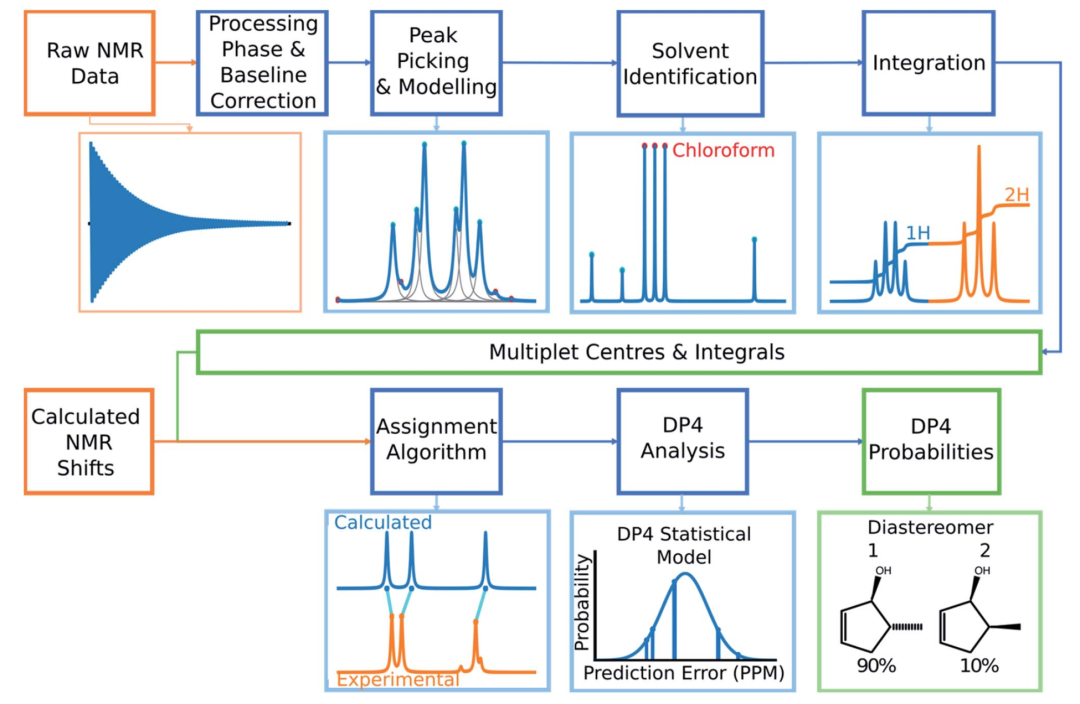
DP4-AI对NMR数据处理的流程如下:当用户输入原始NMR数据后,程序首先对相和基线进行校正,然后从中提取出各个峰的化学位移值,并计算积分值,利用DFT方法计算每个原子的化学位移并对其进行归属,最后DP4分析这种归属的概率,同时给出物质的化学结构。
DP4-AI中NMR峰的提取过程
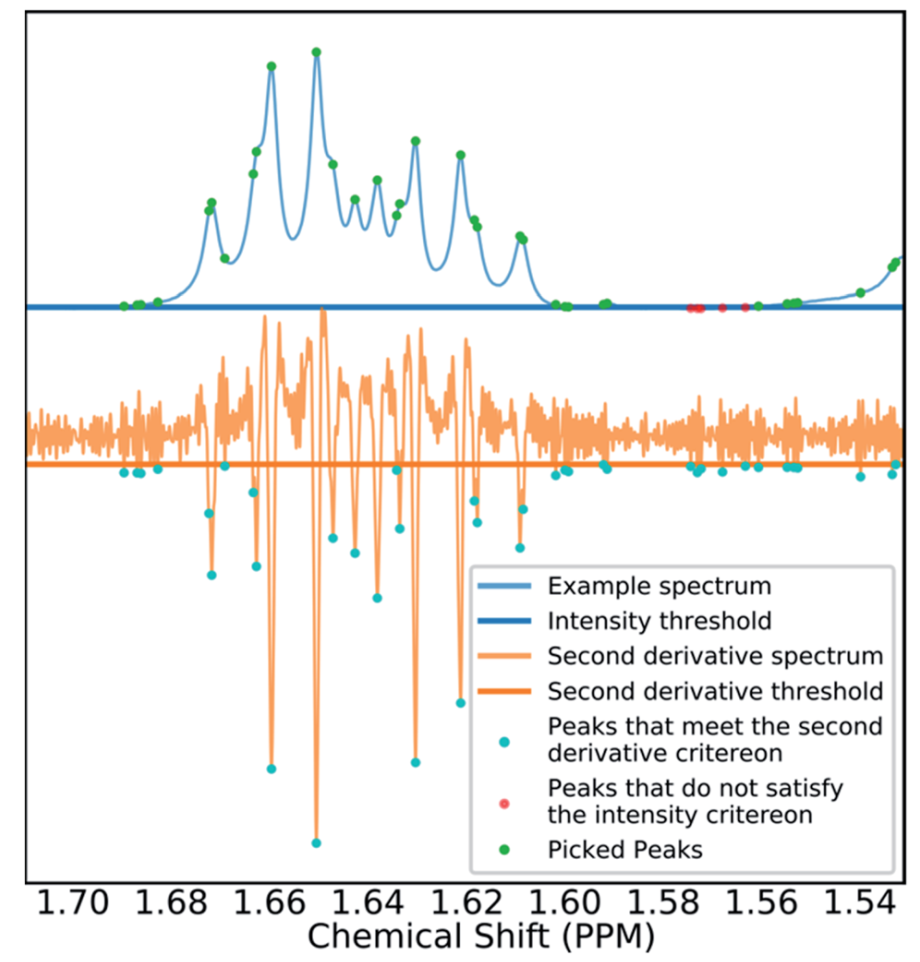
在提取1H NMR位移峰时,使用原始数据的一阶和二阶导数进行操作:如果峰的一阶导数为零,二阶导数最小,而且峰值在二阶导数的幅度阈值以上以及第二阈值以下时,则提取该峰。以这种方式进行峰值提取时可以将两个阈值设置得非常低,在尽可能多地过滤掉噪声的情况下,尽可能少的丢失信号。
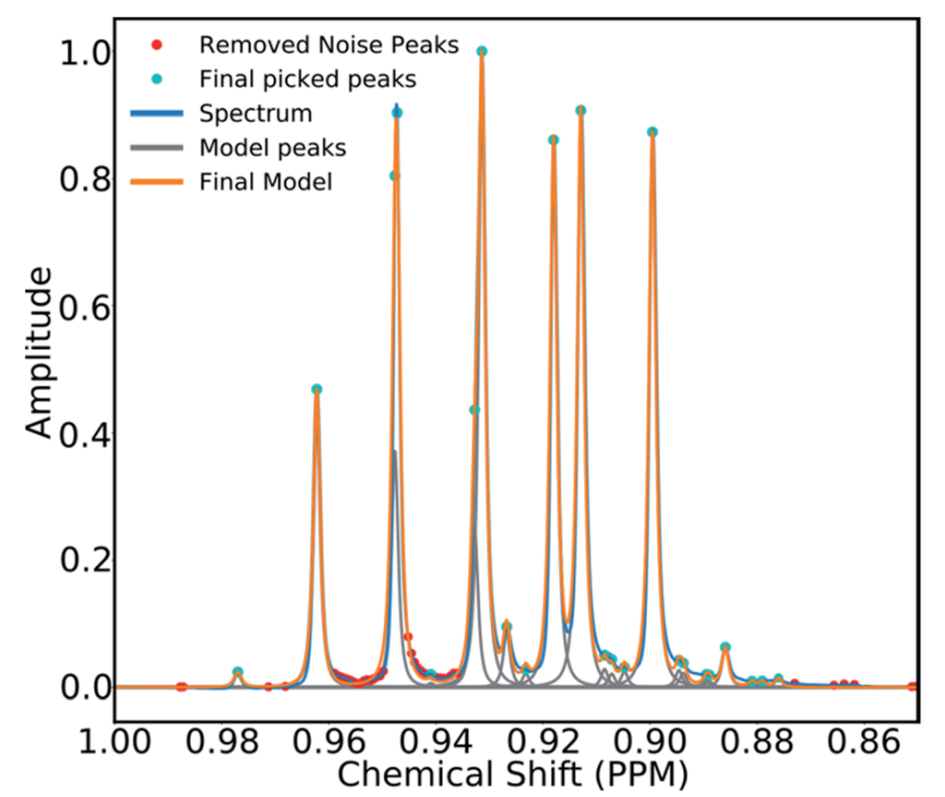
为了避免将噪声误认为信号峰,研究者开发了一种利用目标模型选择来消除噪声的算法。间隔小于18 Hz的提取峰被分组在一起,形成了一个信号区域,对于每个区域,使用多个广义洛伦兹线形函数构建线形模型,每个区域模型中的参数进行迭代变化,直到模型的积分收敛到相应光谱区域1%以内。如果模型的贝叶斯信息低于阈值,则认为这些参数描述的是噪声,相应的峰会被删除。
DP4-AI中NMR峰的归属
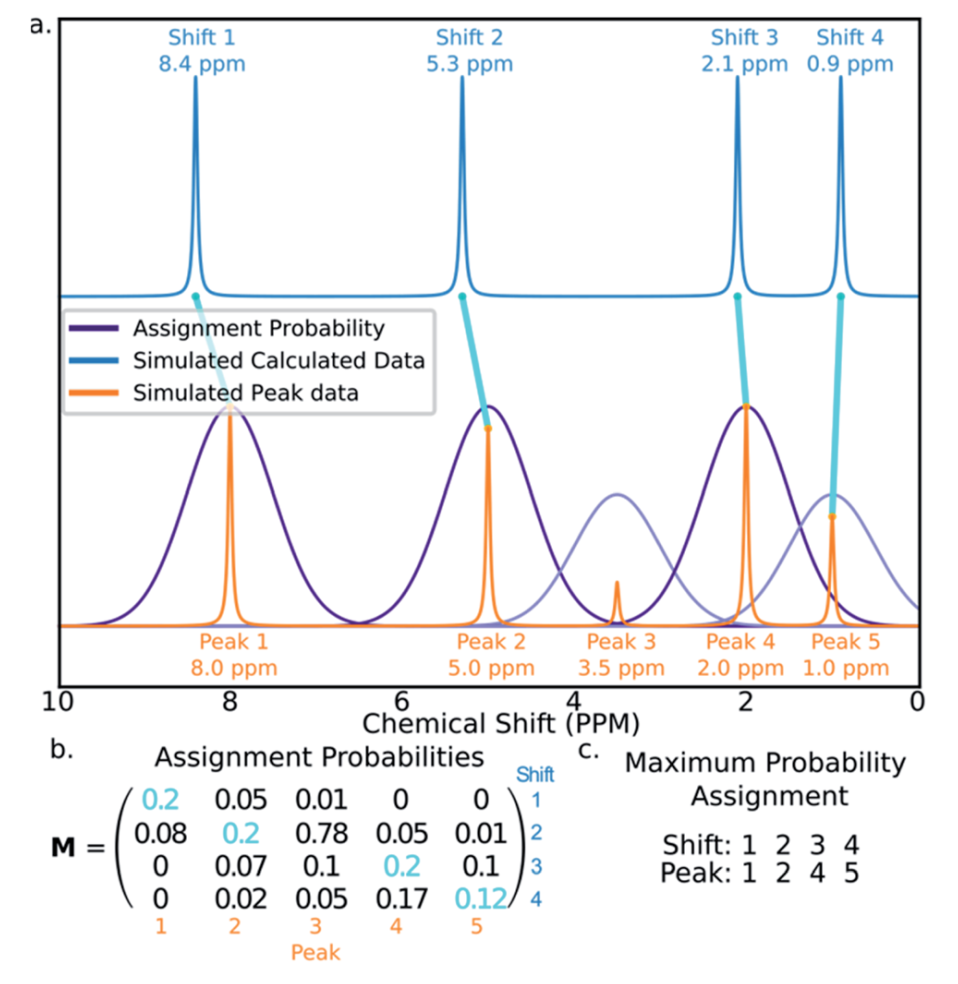
研究者认为DP4-AI开发过程中最具挑战性的工作是归属算法的开发,该算法将分子的每个非对映异构体中的原子分配给光谱中提取到的峰。研究者采用GIAO方法对不同峰进行分配,归属算法的核心是计算分配概率矩阵M,该矩阵的元素Mij是计算出的化学位移i对应于实验峰值j的概率。矩阵M通过匈牙利线性和最小化方法找到最可能的分配结果。
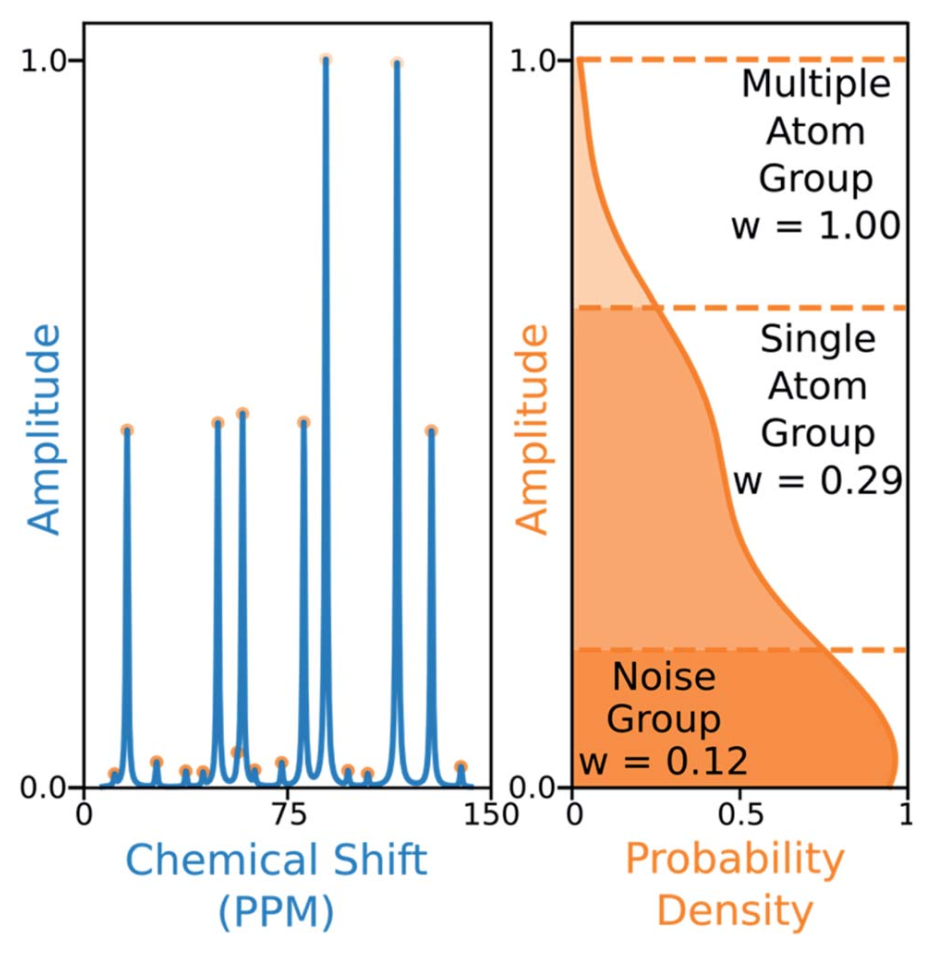
13 C NMR的算法还考虑了实验峰的幅度。M中的每一个元素Mij乘以一个从实验峰值j的幅度得出的权重因子Aj。13 C NMR光谱中的峰通常分为三组,可以通过幅度来区分:噪声、1-原子信号和对应于多个等效碳原子的峰。为了捕获这种变化,研究者估算了谱图中峰值幅度的概率密度函数,当峰值中该函数的二阶导数的最小值位于其振幅之间时,这些峰分为一组,然后使用每组中的峰数和结构中预期的碳原子数来计算振幅权重。
DP4-AI的性能评估
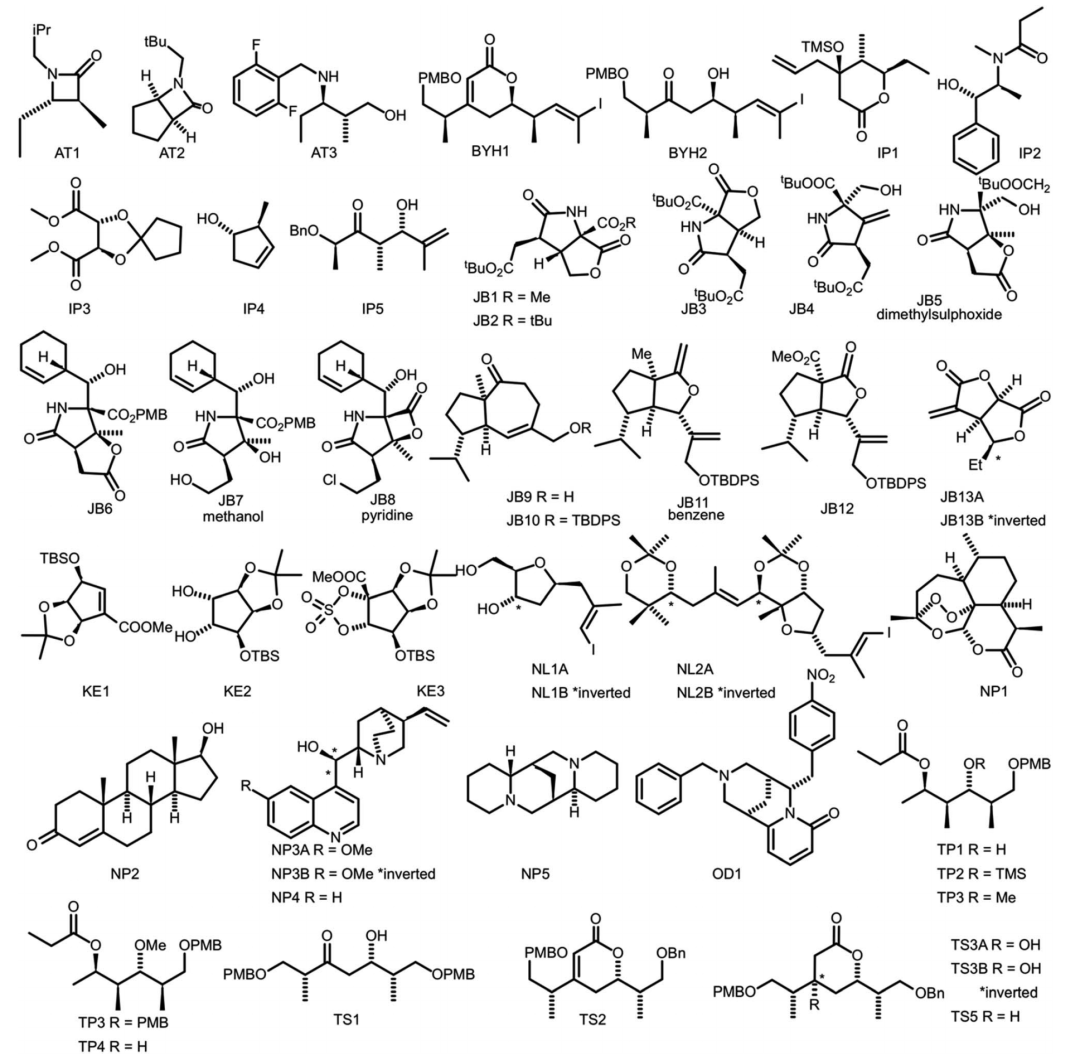
研究者为了评估NMR-AI的性能,构建了由47个分子(每个分子平均3.49个立构中心)组成的测试组,其中包含了各种结构的碳骨架。测试组中包含了天然产物、合成中间体和天然产物的碎片结构,以尽可能包含更多类型的有机分子结构。
为了描述DP4-AI进行NMR预测的误差概率,研究者测试了四个不同的统计模型,结果发现单区域3高斯模型得出的预测误差最优。
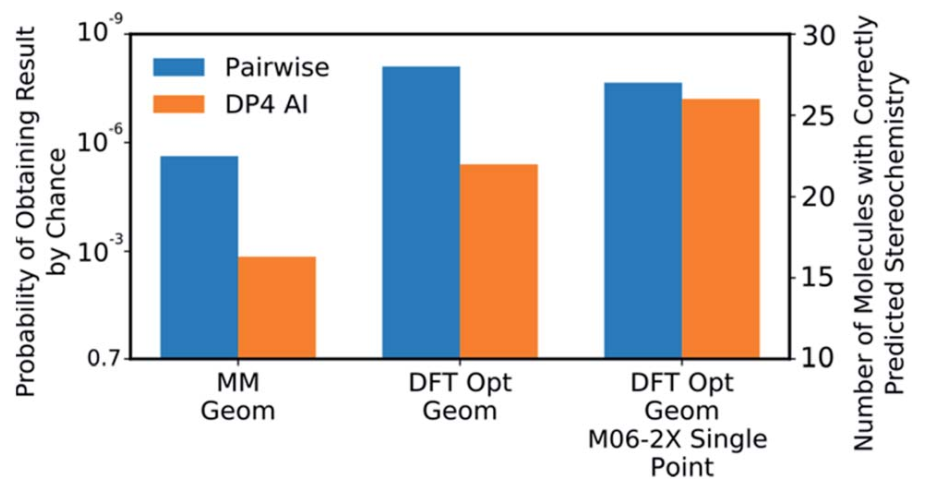
在最高的测试理论水平下,DP4-AI的可靠性与耗时的成对归属算法相似,而后者需要一位训练有素的化学家才能完成。在测试数据集中,正确有效的进行立体化学归属的概率约为3×10-8,表明DP4-AI的表现非常可靠。最令人印象深刻的是,DP4-AI在32和64个非对映异构体中正确对分子NP1和NP2的立构化学特性进行了归属。
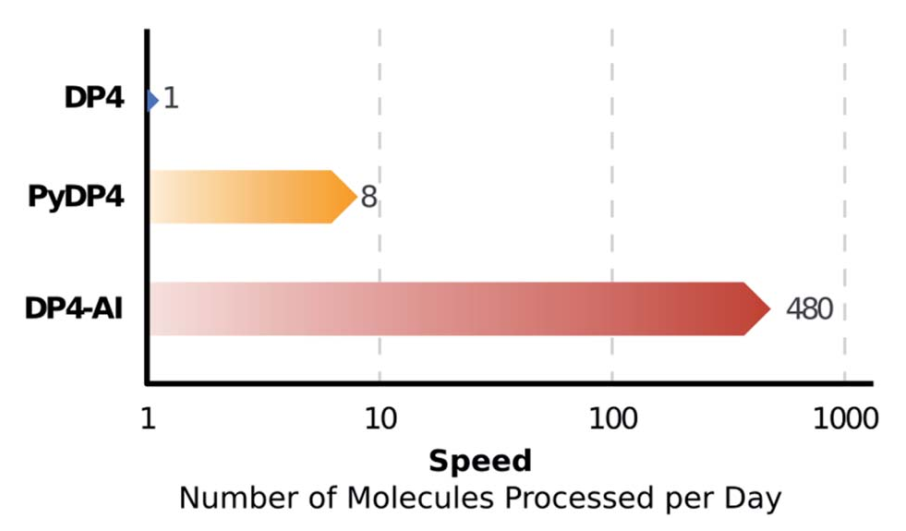
NMR-AI可以在1分钟左右的时间处理完NNR数据,而在此之前同样的任务大约需要8个小时,这相当于每天处理的分子数量可以增加60倍。
小结
为了快速有效的处理NMR原始数据,剑桥大学Jonathan M. Goodman教授课题组提出了一种谱图自动处理和归属方法DP4-AI,这种方法由NMR-AI和PyDP4两部分组成,用户只需要输入原始的NMR数据,程序就会自动提取各个峰值,并对其进行归属,直接给出最可能的分子结构以及这种归属的概率。研究者构建了由47个分子组成的测试组,发现程序正确有效的进行立体化学归属的概率约为3×10-8,并正确对分子NP1和NP2的立构化学特性进行了归属。仅需要1分钟的时间,NMR-AI就可以处理完NNR数据,与之前的方法相比,速率提高了480倍,每天处理的分子数量可以增加60倍。
原文链接:
https://pubs.rsc.org/en/content/articlehtml/2020/sc/d0sc00442a
